

Insulin-related growth factor family: potential role in GIST
Several 2008 papers suggest that the insulin-related growth factors and associated receptors may be treatment targets for GISTs, including wild-type adult GISTs as well as pediatric GISTs and Carney Triad. This is exciting because development of drugs relevant to the insulin-related growth factor (IGF) family is already underway.
Background on IGF Family
The IGF family includes two growth factors (IGF-1 and IGF-2, sometimes called IGF-I and IGF-II), two receptors (IGF-1R and IGF-2R), and six binding proteins (IGFBP1-6) that modulate the actions of IGFs by keeping them bound in the bloodstream and therefore unavailable. The receptor IGF-1R can be activated by either IGF-1 or by IGF-2 to result in cell growth (proliferation) and resistance to cell death (apoptosis). The receptor IGF-2R does not trigger signaling, but rather serves to take IGF-2 out of circulation.
In normal physiology the IGF family is involved in childhood growth, puberty, pregnancy, and lactation (breast milk production). IGF-1 is produced primarily by the liver as an endocrine hormone (circulating in the bloodstream), but it is also produced in target tissues in a paracrine/autocrine fashion for local signaling. Most body cells express the receptor IGF-1R and can be affected by IGF-1, especially cells in muscle, cartilage, bone, liver, kidney, nerves, skin, and lungs. In addition to the insulin-like effects, IGF-1 can also regulate cell growth and development, especially in nerve cells, as well as cellular DNA synthesis. IGF-1 is produced throughout life. The highest rates of IGF-1 production occur during the pubertal growth spurt. The lowest levels occur in infancy and old age. (see wikipedia pages on IGFR-1 and IGF-1.
The IGF family is closely related to the insulin signaling system (insulin and the insulin receptor) that controls metabolism. In fact, IGF-1R can be activated by insulin. Hybrid receptors also exist that are a combination of half insulin receptor and half IGF-1R; these can be activated either by insulin or by IGF-1 or IGF-2.
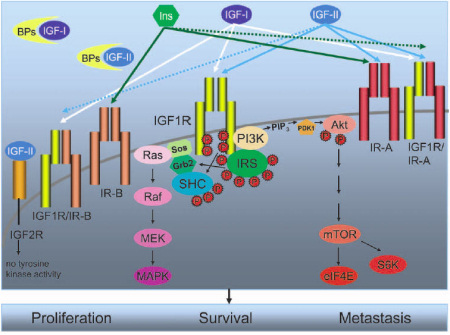
Figure 1. The IGF system components. This figure is reproduced with the kind permission of the authors and Molecular Cancer Therapeutics from the paper by Sachdev and Yee (2007) in Molecular Cancer Therapeutics 6(1): 1-12.
The IGF system is composed of three ligands: IGF-I, IGF-II, and insulin. IGF-I and IGF-II are found in the circulation complexed to a family of binding proteins, called IGF binding proteins (BPs), which serve to regulate bioavailability of these ligands in the tissues. There are multiple receptor conformations. IGF-IR is a transmembrane tyrosine kinase and consists of two alpha-beta chains and has high affinity for both IGF-I and IGF-II. The insulin receptor (IR) mediates the effects of insulin. Two isoforms of IR, IR-A and IR-B, are found in breast cancer cells, with IR-A being overexpressed in many cancers. In addition, hybrid receptors assembled with one alpha-beta chain of IGF-IR (IGF1R) and one alpha-beta chain of IR-A or IR-B also exist. Arrows, ligand specificity of the various receptors; solid arrows, high-affinity binding; dotted arrows, low-affinity binding. Thus, IGF-I binds to IGF-IR and IGF-IR/IR-B with high affinity (solid white arrows) and to IGF-IR/IR-A hybrid receptor with much reduced affinity (white dotted arrow). IGF-II binds to IGF-IR, IR-A, and hybrid IGF-IR/IR-B and IGF-IR/IR-A receptors. For simplicity, only signaling initiated by the activated IGF-IR is shown. Activation of IGF-IR results in phosphorylation of adaptor proteins belonging to the IRS family or SHC. Activation of IRS and SHC leads to activation of extracellular signal—regulated kinase (ERK) 1/2 of the MAPK cascade via the growth factor receptor binding protein 2 (Grb2)/Sos/Ras/Raf/MAPK extracellular signal—regulated kinase kinase (MEK) pathway. IRS proteins also bind to the p110 subunit of phosphatidylinositol 3 ¶-kinase (PI3K), leading to the generation of phosphatidylinositol3,4.5-triphosphate (PIP3) and phosphorylation of Akt by phosphoinositide dependent kinase (PDK1). Phosphorylation of Akt leads to subsequent activation of mammalian target of rapamycin mTOR), eukaryotic translation initiation factor 4E (eIF4E), and p70S6 kinase (S6K). Activation of these downstream signaling pathways leads to enhanced proliferation, survival, and metastasis in cancer cells. Similar signaling pathways are activated by IR and the various hybrid receptors shown.
Several different types of cancer have previously demonstrated activity of the IGF family, which serves both to increase cancer cell proliferation (cancer growth), and to inhibit cancer cell apoptosis (cell death). IGF pathway activation can reduce the ability of some cancer drugs to cause apoptosis. IGF-1R is also involved in cell motility and anchorage-independence (the ability of cancer cells to leave the tumor and travel in the bloodstream to cause metastasis). Loss of IGF-2R in tumors effectively increases signaling by IGF-2.
IGF family signaling has been implicated in several types of sarcomas (Magenau and Scheutze, 2008, PMID 18525335; Scotlandi and Picci, 2008, PMID 18525338). Phase II trials of IGFR-1 inhibitors are in progress for Ewing’s sarcoma, osteosarcoma, and rhabdymyosarcoma. Side effects may be minimal as long as inhibition of the structurally similar insulin receptor can be avoided; otherwise a diabetic condition could result. The existence of "hybrid" receptors complicates drug targeting. Alternate strategies include targeting IGF-1 and IGF-2 instead of the receptor, as discussed by Sachdev and Yee, 2007 (PMID 17237261). For a free-access review on targeting the IGF pathway see Ryan and Goss, 2008 (PMID 18245009).
Italian Research Findings About IGF Family in GIST
Italian researchers Braconi et al (PMID 18372285) assessed the expression of IGF-1 and IGF-2 in 94 KIT-expression-positive adult GIST samples using immunohistochemistry (cell staining methods). They scored IGF-1 and IGF-2 expression as negative, moderate, or strong. The results showed a relationship of strong IGF-1 expression to higher tumor risk category, larger tumor size, higher mitotic rate, and to the presence of metastases at diagnosis. Strong IGF-2 expression was related to higher risk category and higher mitotic rate. Follow-up data on the 77 patients without metastases at diagnosis showed relapse in 18 cases. Relapse was significantly more likely in patients whose tumors had higher expressions of either IGF-1 or IGF-2 or both. Among high-risk tumor patients, there was a trend for reduced recurrence rate in patients without expression of either IGF-1 or IGF-2. Interestingly, in this study 100% of GISTs expressed the receptor IGF-1R in the cell cytoplasm.
Braconi et al hypothesized that IGF-1R activation could trigger the MAPK pathway independently of KIT activation, and that GISTs could execute a self-sustaining autocrine loop of IGF activation causing GIST proliferation and preventing GIST cell death. (Autocrine means that a single cell both produces the sign
al and receives it: self-stimulation.) Of potential importance for patients with KIT exon 9 mutation, IGF-2 expression was strong in 70% of the exon-9 GISTs in this study. In contrast, in the less aggressive tumors with PDGFRA mutations, the majority (64%) were negative for IGF-2 expression. IGF-1 expression did not differ by mutation type.
MSKCC Research Results About IGFR-1 in Pediatric GISTs
Cristina Antonescu and a team from Memorial Sloan-Kettering Cancer Institute (Agaram et al, PMID 18483389) examined tissue from 17 pediatric GIST patients and compared genes expressed in the pediatric cases with the genes expressed in adult wild-type GIST and adult gastric GISTs. The MSKCC team found that IGF-1 expression was five times higher in pediatric GIST than in adult wild-type GIST based on microarray analysis of transcriptional profiles. The receptor IGF-1R was likewise overexpressed. [Other gene expression results in this study, as well as drug testing results on cell lines, are not summarized here because they are unrelated to IGF family topics.]
Fox Chase Findings on IGF in Wild-Type and Mutant GISTs
Andrew Godwin and his lab team investigated IGF signaling in wild-type GISTs (including both a GIST and a paraganglioma from one Carney Triad patient) compared to KIT-mutant GISTs (Tarn et al, 2008, PMID 18550829). In addition to the new published paper also see the presentation by Godwin at 2008 ASCO, abstract number 10507). Consistent with the Italian findings, the Fox Chase team identified IGF-1R expression in all GISTs, but especially in wild-type GISTs. The Fox Chase team detected gene copy number changes for the IGF-1R gene using microarrays and found that 70% of wild-type GISTs showed amplification of IGF-1R (2.5 to 4 copies), as did 27.7% of KIT-mutant GISTs. Clinically convenient cell staining techniques (immunohistochemistry) confirmed these results.
Next the investigators evaluated the effect of an anti-IGF-1R drug (NVP-AEW541) on two GIST cell lines that differed in sensitivity to imatinib (T1 cells with an exon 11 KIT mutation and greater sensitivity to imatinib, and 882 cells with an exon 13 KIT mutation and reduced sensitivity to imatinib). Both cell lines were equally sensitive to the IGF-1R inhibitor. Moreover, there was an additive effect of combining imatinib plus NVP-AEW541 on achieving cell death. Because using interfering RNA to silence IGFR-1 had less effect than NVP-AEW541, the researchers suspected that the drug also produces additional effects besides IGF-1R inhibition. Interestingly, consistent with the Italian hypothesis that IGF-1R activation could trigger the MAPK pathway independently of KIT activation, drugging IGF-1R did particularly reduce MAPK path activation.
The Fox Chase team concluded that drug treatment to inhibit IGF-1R needs to be investigated as a potentially useful strategy in GIST, both
- as a single agent in tumors that do not respond well to imatinib (such as adult wild-type tumors and pediatric GIST), and
- as a combined therapy to increase cell death in imatinib-responsive tumors.
Investigational Drugs Against the IGF Family
Several drugs targeting IGF family are in clinical trials at present or expected to enter trials, including the following:
- CP-751,871
- R1507 (see NCT00642941 trial for sarcomas)
- AMG 479
- OSI906 (see NCT00514007 and NCT00514306)
- NVP-AEW541
Speculative Remarks
If further research were to confirm the Italian finding that exon-9-mutant GISTs showed especially strong expression of IGF-2, then targeting the IGF family in exon-9 GIST patients might help improve their response to Gleevec.
It is conceivable that in adult GISTs with KIT mutations, IGF-1 or IGF-2 expression might interfere with tumor cell response to imatinib by preventing apoptosis. Recently the epidermal growth factor receptor (EGFR) was found to prevent apoptosis in cancer cells which did NOT depend on EGFR signals for growth (Fidler et al, 2008, PMID 18455122.) If this were true, then combining an inhibitor of the IGF family with a KIT inhibitor (such as imatinib) might assist in killing more GIST cells and improving treatment response.
The normal occurrence of high IGF-1 levels during puberty and pregnancy certainly fits in with a potential link to pediatric GIST and Carney Triad tumors, which typically occur at these life stages. IGF signaling is to be a topic of discussion at the first NIH Pediatric GIST Clinic being held June 18-20, 2008, and we will be eager to hear the ideas from that meeting.
IGF-1 can activate hypoxia-indicible factor (HIF) pathways (Carroll and Ashcroft, PMID 16778202), which are relevant to pediatric GIST. In pancreatic cancer cells, HSP90 inhibition impaired IGF signaling as well as HIF pathways and reduced growth of tumors transplanted into mice (Lang et al, 2007, PMID 17975158). Therefore, it is possible that drugs to inhibit HSP90 and IGF pathways might work well together.
Trent et al (2006, PMID 16986125) found that when imatinib (Gleevec) is effective early in GIST treatment, the binding protein IGFBP-3 is upregulated; conversely, in imatinib-resistant GIST cells IGFBP-3 was down-regulated. The authors stated "The increase we found in IGFBP3 in imatinib-sensitive GIST cells and tumor tissue suggests that this protein may be an important early marker of FDG-PET imaging and the antitumor activity of imatinib." however, Trent et al did not offer any hypotheses about exactly how imatinib (Gleevec) could be affecting IGFBP-3.
Perhaps soon the relationships between different IGF family elements and GIST growth will be unravelled and will provide an effective treatment approach.
Additional Reading:
For free-access reviews about the insulin-related growth factor system in cancer, see these papers:
Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK
Insulin-like growth factor 1 receptor (IGF-1R) is a potential therapeutic target for gastrointestinal stromal tumors (GIST).
Proc Natl Acad Sci U S A. 2008 Jun 17;105(24):8387-92.
PMID: 18550829
Ryan PD, Goss PE.
The emerging role of the insulin-like growth factor pathway as a therapeutic
target in cancer.
Oncologist. 2008 Jan;13(1):16-24.
PMID 18245009.
Sachdev D, Yee D.
Disrupting insulin-like growth factor signaling as a potential cancer therapy.
Mol Cancer Ther. 2007 Jan;6(1):1-12.
PMID 17237261;
Samani AA, Yakar S, LeRoith D, Brodt P.
The role of the IGF system in cancer growth and metastasis: overview and recent insights.
Endocr Rev. 2007 Feb;28(1):20-47.
PMID 16931767
Yu H, Rohan T.
Role of the insulin-like growth factor family in cancer development and progression.
J Natl Cancer Inst. 2000 Sep 20;92(18):1472-89. Review.
PMID 10995803;